
Le Amiloidosi – Classificazione
Le amiloidosi costituiscono un gruppo eterogeneo di disordini del metabolismo proteico accomunati dal deposito in sede extracellulare di fibrille proteiche insolubili che si colorano positivamente con...
Le amiloidosi costituiscono un gruppo eterogeneo di disordini del metabolismo proteico accomunati dal deposito in sede extracellulare di fibrille proteiche insolubili che si colorano positivamente con...

Le cellule fagocitarie (neutrofili, monociti) assieme alle cellule natural killer, al complemento e altre proteine plasmatiche concorrono a formare il sistema immunitario innato, evolutosi e perfez...

Oggi conosciamo almeno 31 precursori proteici delle fibrille amiloidi, molti dei quali circolano liberamente nel plasma. i vari tipi di amiloidosi possono manifestarsi come malattie sistemiche e gener...

La sindrome di Richter è una rara complicanza che compare nel 5-10% dei pazienti con leucemia linfatica cronica [1,2]. La SR consiste nello sviluppo di linfoma aggressivo diffuso grandi cellule B che...

La talidomide è una molecola derivata dall'acido glutammico dotata di proprietà immunomodulanti e antinfiammatorie [1]. Assieme ad alcuni suoi derivati, anch’essi somministrabili per via orale, s...

La sarcoidosi è una malattia infiammatoria granulomatosa sistemica di origine sconosciuta. Il coinvolgimento del sistema nervoso, o neurosarcoidosi, può presentarsi in qualsiasi regione del sistema...

Nel 3-10% dei casi la leucemia linfatica cronica (LLC-B) può trasformarsi in un linfoma a maggiore aggressività istologica e clinica, evento noto anche come sindrome di Richter (SR). Nella versione...

La variante di Hodgkin della sindrome di Richter, cioè la trasformazione il linfoma di Hodgkin della LLC-B è un'evenienza molto rara. Il termine "variante di Hodgkin della sindrome di Richter" è st...
La sindrome di Richter è una rara complicanza che compare nel 5-10% dei pazienti con leucemia linfatica cronica [1,2]. La SR consiste nello sviluppo di linfoma aggressivo diffuso grandi cellule B che...

I granulociti neutrofili o granulociti polimorfonucleati (PMN) fanno parte del sistema immunitario innato e rappresentano una prima linea di rapida difesa dell’ospite contro le infezioni; partecipan...

La sarcoidosi è una malattia infiammatoria granulomatosa sistemica di origine sconosciuta. Il coinvolgimento del sistema nervoso, o neurosarcoidosi, può presentarsi in qualsiasi regione del sistema...

La storia naturale della neurosarcoidosi è scarsamente definita. Non sono stati finora condotti studi clinici randomizzati, in doppio cieco e controllati contro placebo sulla terapia della neurosarco...

La chemioimmunoterapia (R-CHOP, R-EPOCH e simili) e i nuovi farmaci diretti contro bersagli molecolari intracellulari hanno dimostrato una ridotta efficacia nella sindrome di Richter (SR). Risposte c...

La sarcoidosi è stata descritta in tutti i gruppi etnici e in tutte le fasce di età di entrambi i sessi. La sua incidenza tuttavia varia secondo l’età, il genere, l’etnia e l’origine dei paz...
In seguito alla diffusione su larga scala dei contaglobuli elettronici, è aumentata notevolmente la frequenza con la quale il medico deve valutare casi di linfocitosi assoluta clonale persistente in...
La presenza anche di minime quantità di linfociti B monoclonali in soggetti apparentemente normali e senza linfocitosi assoluta è nota come linfocitosi monoclonale delle cellule B e diversi altri si...

L’ECG standard, l’Holter, l’ecocardiografia, la scintigrafia miocardica con radionuclidi, la risonanza magnetica nucleare, la TAC, la PET, sono le metodiche non invasive preferite per la valutaz...

La sarcoidosi cardiaca, o cardiosarcoidosi, è una rara localizzazione della sarcoidosi, una malattia granulomatosa ad eziologia sconosciuta le cui caratteristiche lesioni, i granulomi non caseos...

La sarcoidosi è una malattia infiammatoria granulomatosa cronica ad eziologia sconosciuta, caratterizzata dalla presenza di granulomi non caseosi, non necrotizzanti, e dall’accumulo di linfociti T ...
Lo stomaco è il segmento del tratto gastrointestinale più frequentemente interessato, seguito dall’esofago, appendice, colon, retto, pancreas e peritoneo. La sarcoidosi GI si manifesta con sintomi...

La mononucleosi infettiva (MI) è una sindrome clinica caratterizzata dalla triade sintomatologica febbre, faringite e linfoadenopatia. L’EBV o virus di Epstein Barr è la causa più frequente di MI...

Le gammapatie monoclonali di incerto significato o MGUS, Monoclonal Gammopathy of Undetermined Significance, sono caratterizzate dalla presenza di una componente monoclonale nel siero e/o nelle urine....

Il mieloma multiplo (MM) è una neoplasia maligna delle plasmacellule caratterizzata da aumento delle plasmacellule midollari e dalla presenza di una componente monoclonale nel siero e/o nelle urine. ...

Il deficit di granuli specifici dei neutrofili è una rara malattia caratterizzata da anomalie morfologiche e funzionali dei granulociti neutrofili caratterizzata da riduzione del numero dei granuli...

I pazienti con MGC presentano, spesso a partire dalla prima infanzia, infezioni ricorrenti batteriche e fungine rappresentate in ordine di frequenza da polmoniti, ascessi (cute, tessuti, organi inter...

La malattia granulomatosa cronica (MGC) comprende un gruppo eterogeneo di disordini genetici caratterizzati clinicamente da gravi infezioni ricorrenti batteriche e fungine e dall’anomala formazione...

Le pietre miliari del trattamento dei pazienti con MGC sono: la diagnosi precoce per facilitare la profilassi antimicrobica e immunomodulatrice e il trattamento aggressivo e precoce delle infezioni. ...

Le cellule fagocitarie (neutrofili, monociti) assieme alle cellule natural killer, al complemento e altre proteine plasmatiche concorrono a formare il sistema immunitario innato, evolutosi e perfez...

La triade costituita da insufficienza pancreatica esocrina, neutropenia, anomalie scheletriche, è nota come sindrome di Shwachman-Diamond (SDS), trasmessa come carattere autosomico recessivo. Fra le ...
La triade costituita da insufficienza pancreatica esocrina, neutropenia, anomalie scheletriche, è nota come sindrome di Shwachman-Diamond (SDS), trasmessa come carattere autosomico recessivo. Il gene...

La carenza di glucosio-6-fosfato deidrogenasi (G6PD), un deficit della via dello shunt dell'esoso monofosfato è il'anomalia più frequente del metabolismo dei globuli rossi a livello gloale. Il gene...

Nella letteratura medica il termine "neutropenia congenita" è utilizzato con diversi significati a volte causa di confusione. "Congenito" significa letteralmente una riduzione del numero dei neutrofi...

Prima dell'avvento del G-CSF ricombinante, i pazienti con NCG erano convenzionalmente trattati con antibiotici o solo durante gli episodi infettivi o come profilassi per aumentare il numero dei neutro...

La neutropenia ciclica è una rara malattia caratterizzata da episodi recidivanti di febbre e stomatite aftosa della durata di 3-7 giorni e la cui comparsa avviene in corrispondenza di ricorrenti per...
Già al momento della nascita, o poco dopo, i pazienti si con infezioni gravi e ricorrenti a comparsa nel periodo neonatale o infantile. Nei neonati la modalità di presentazione è spesso un’infe...

La leucocitosi con aumentato numero di bastoncelli (cioè spostamento a sinistra della formula di Arneth) è l’usuale risposta dell’organismo alle infezioni. Alcune infezioni come la febbre tifoid...

Le neoplasie plasmacellulari, o discrasie plasmacellulari, costituiscono un gruppo eterogeneo di sindromi causate dalla proliferazione di un clone di linfociti B maturi che mantiene la capacità di d...
Il plasmocitoma solitario è un tumore singolo composto da plasmacellule monoclonali istologicamente identiche a quelle osservate nel mieloma multiplo, in assenza di sintomi e segni di altre localizz...

La progressione a mieloma multiplo compare nel 50-60% dei pazienti con PSO durante il decorso della loro malattia. Numerosi fattori prognostici sono stati associati alla prognosi al momento della ...

La terapia del PSO si basa principalmente sulla radioterapia locale a dosi adeguate; il ruolo della chemioterapia adiuvante e nei casi ad alto rischio è controverso, mentre la chirurgia ha un’impor...

La progressione a mieloma multiplo compare nel 50-60% dei pazienti con PSO durante il decorso della loro malattia. Numerosi fattori prognostici sono stati associati alla prognosi al momento della ...

Il mieloma asintomatico, o “smoldering”, è una neoplasia plasmacellulare che non ha ancora provocato danni d’organo clinicamente evidenti ma ha un elevato rischio di progressione a mieloma clin...
Le sindromi da iper IgE sono caratterizzate da infezioni ricorrenti, principalmente a carico della cute e delle vie aeree superiori ed inferiori, elevati livelli nel siero di IgE, anomalie di altri o...
Il deficit di Dedicator of Cytokinesis number 8 (DOCK-8) (OMIM*243700) o hyper-IgE Syndrome 2 (HIES-2) è una sindrome autosomica recessiva condivide con la HIES-1 l’aumento delle IgE, l'eosinofil...

ZNF341 è un fattore di trascrizione nucleare di tipo zinc finger che si lega al promoter di numerosi geni, fra i quali STAT3. Mutazioni Loss of Function eterozigoti di ZNF341, un gene precedentemente...

I deficit di fosfoglucomutasi 3 (PGM3) causano una forma di sindrome da iper-IgE (HIES) a trasmissione autosomica recessiva (AR). I deficit congeniti di PGM3 e di altri enzimi coinvolti nella glicos...

Il deficit di IL6ST può essere causato da mutazioni omozigoti o eterozigoti composite del gene IL6ST, che codifica per la gp130, oppure da mutazioni eterozigoti dominanti negative di un solo allele d...

Il deficit di TYK2 causa una rara sindrome da immunodeficienza primitiva con predisposizoine alle infezioni da micobatteri, da altri batteri intracellulari e virali. La sindrome da iper-IgE, descritt...
ILgene IL-6R codifica per la subunità alfa del recettore per IL-6..L‘altra subunità del recettore, la gp130 o IL-6ST, permette la trasmissione del segnale originato dall’attivazione di IL-6R all...
La sindrome di Comèl-Netherton (NETH, OMIM*25650) è una rara malattia autosomica recessiva con interessamento della pelle, dei capelli del sistema immunitario caratterizzata dalla triade di ittiosi...
La sindrome di Loeys-Dietz ( LDS ) è una malattia genetica a trasmissione autosomica dominante con manifestazioni cliniche prevalenti a carico del tessuto connettivo e dell’apparato vascolare. Nei ...

Le mutazioni omozigoti null di CARD11 causano un’immunodeficienza combinata grave (SCID) a trasmissione autosomica recessiva (AR), caratterizzata da: normale numero di linfociti B e T, ma con defi...

La risposta di difesa dell’organismo ai microrganismi esterni è il risultato del concorso di un insieme di sistemi di protezione, comprese barriere fisiche, vari tipi cellulari e mediatori solubili...

I leucociti migrano dal circolo al sito dell’infiammazione seguendo un gradiente di chemochine provenienti dalla sede dell’infiammazione/infezione in un processo noto come chemiotassi. I fattori c...

La LAD-3, (OMIM 612840), è causata da mutazioni del gene FERMT3 (o KINDLIN3) (OMIM*607901) che codifica per una proteina intracellulare, la Kindlina 3, che interagisce con le β2 integrine. Oltre a...
La LAD-2 è causata da mutazioni nel gene SLC35C1 (OMIM*605881) (Solute carrier family 35 member C1), che è localizzato sul cromosoma 11p11.2. SLC35C1 codifica per la proteina GDP-fucosio transpor...

Le immunodeficienze combinate (CID) sono delle rare sindromi da errori congeniti dell’immunità caratterizzate da gravissimi deficit quantitativi e/o qualitativi dei linfociti T con o senza difetti...

I sintomi tipici della SCID sono: le gravi infezioni ricorrenti, soprattutto delle vie respiratorie e del tratto gastrointestinale; la diarrea cronica e la ridotta velocità di crescita. Se non ident...

L’immunodeficienza combinata severa (Severe Combined Immune Deficiency, SCID) legata al sesso o X-linked è la forma più frequente fra tutte le immunodeficienze primitive combinate. È nota anche...

Il deficit selettivo o isolato di IgA è il deficit immunitario più frequente a livello mondiale. È solitamente asintomatico e non necessita di terapia alcuna. I pochi soggetti che manifestano infez...

Il 90 % degli individui con deficit isolato di IgA è asintomatico, come è dimostrato dalla sua prevalenza in donatori sani. Il resto dei pazienti è predisposto a sviluppare infezioni ricorrenti de...

La SCID T-B-NK+ SCID è causata nell’uomo da mutazioni di almeno 5 geni diversi coinvolti nella ricombinazione V(D)J dei recettori di membrana linfocitari, le immunoglobuline (Ig) nei linfociti B ...

CD45 è codificata dal gene PTPRC (Protein Tyrosine Phosphatase Receptor Type C). CD45 esiste in diverse isoforme, originate da splicing alterativo degli esoni 4,5, 6. CD45 è una molecola recettorial...
Il deficit di beta-2 microglobulina è stato descritto finora in soli due casi che presentavano una grave immunodeficienza di tipo combinato o SCID..
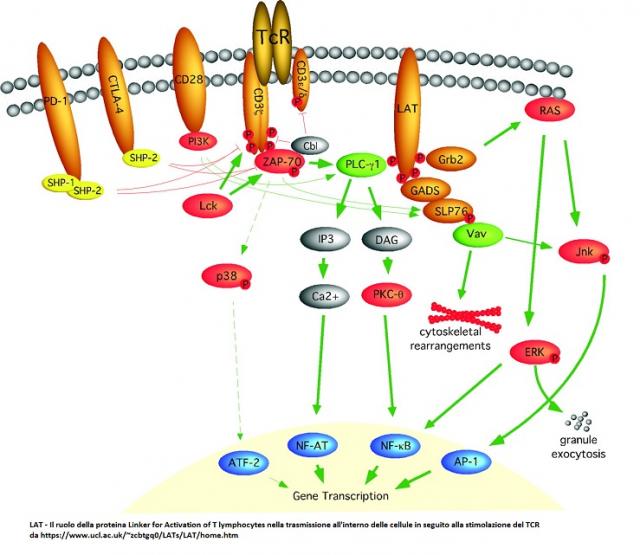
Il legame fra TCR e un suo ligando, un antigene peptidico presentato da una proteina del complesso maggiore di istocompatibilità, innesca cascate di eventi biochimici il cui esito è la stimolazione...

Fra le sette coronine note, la 1a è la più importante nei mammiferi e anche quella più studiata. Le mutazioni autosomiche recessive del gene CORO1A causano nei topi e nell'uomo linfocitopenia T di...

Il deficit di molecole MHC II generalmente si traduce in un quadro clinico di SCID, indipendentemente dal difetto molecolare, poiché esse svolgono un ruolo fondamentale nella maturazione e nella f...

Il deficit di IL-7Rα causa un deficit selettivo dei linfociti T ed è una rara causa di SCID T-B+NK. Solo pochi casi sono sstati segnalati, in genere in bambini nati da genitori consanguinei. Non esi...

I linfociti T maturi riconoscono gli antigeni per mezzo di un eterodimero variabile (αβ o γδ), della membrana di superficie noto come TCR o recettore delle cellule T. Nell'uomo, le subunità d...

L'anomalia peculiare della SCID è la linfocitopenia, solitamente <1500/µL, che non è però costante [6]. Tuttavia, alla nascita numero assoluto di linfociti <2000/µL, deve far sorgere il sospe...

I linfociti T maturi hanno due tipi di recettore di membrana per l’antigene o T cell Receptor (TCR) : TCR2 o TCRa-b presente nel 95% dei linfociti T periferici e costituito da una catena a e da un...

Cernunnos, dal nome di una divinità celtica protettrice della fertilità, noto anche come Non-homologous end-joining factor 1 (NHEJ1) è un fattore di riparazione del DNA essenziale per il processo d...

Il gene DNA ligasi IV, o LIG4, proteina essenziale per la giunzione terminale non omologa del DNA, nonhomologous DNA end joining (NHEJ), il processo che opera il ricongiungimento delle estremità del...

Il gene DNA cross-link repair 1C (DCLRE1C) è indispensabile per la ricombinazione dei geni variabili V(D)J dei recettori TCR e BCR sui linfociti T e B e per lo svolgimento della NHEJ che permette la...

Le mutazioni del gene Protein Kinase, Dna-Activated, Catalytic Subunit (PRKDC ) (OMIM*600899) sono trasmesse come carattere autosomico recessivo e sono causa di una forma di SCID T-B-NK+

Le Neutropenie croniche gravi (NCG) sono considerate condizioni preleucemiche nelle quali l’ottenimento di un chimerismo completo, cioè la totale sostituzione delle cellule staminali emopoietiche d...

I farmaci citotossici tradizionali raramente inducono anomalie tiroidee in assenza di irradiazione. Tuttavia, possono sensibilizzare la tiroide agli effetti della concomitante radioterapia aumentando ...

Riassunto Mutazioni del gene adenosina deaminasi causano accumulo substrati e metaboliti tossici per le cellule, fra quali adenosina, desossiadenosina e desossi-ATP che inducono danni cromosomici b...

Riassunto Mutazioni del gene PNP (OMIM*164050) causano accumulo substrati e metaboliti tossici per le cellule. soprattutto per quelle del sistema immunitario, fra quali adenosina, desossiadenosina...

Il deficit di adenilato chinasi 2 (AK2) causa la disgenesia reticolare, un’immunodeficienza combinata grave di tipo SCID T-B-NK- associata ad agranulocitosi e sordità neurosensoriale bilaterale....

Le sindromi da iper IgM sono un gruppo di rare immunodeficienze primitive congenite ed ereditarie caratterizzate da: infezioni ricorrenti ad insorgenza precoce, diminuzione o assenza nel siero di IgG ...

Il gene IkKgamma (NEMO) codifica per una proteina che, assieme a due altre subunità denominate Ikalfa e IkKbeta, formano il complesso enzimatico IkBK (IKK). IkBK è una chinasi che catalizza la fos...

Il Nuclear Factor-kappa B (NF-kB) è un fattore di trascrizione ubiquitario conservato nelle cellule eucariotiche. NF-kB è attivato da numerosi stimoli compresi prodotti virali e batterici, radiazio...

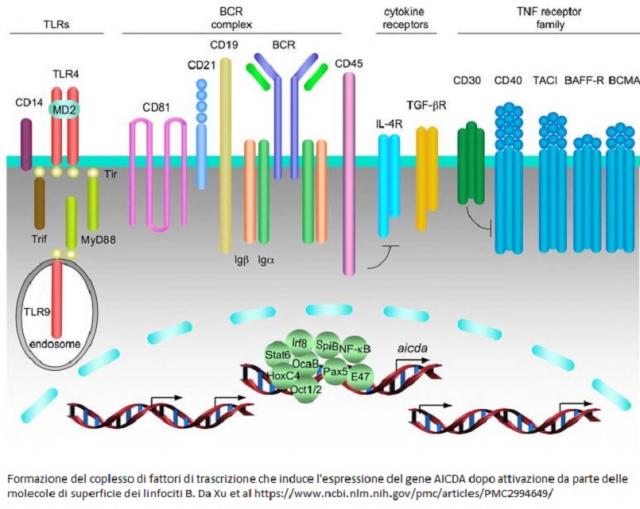
Il deficit di adenosina deaminasi indotta dall'attivazione (AICDA) è caratterizzato da bassi o assenti livelli nel siero di IgG, di IgA e di IgE, con normali o elevati livelli di IgM. I pazienti aff...

Il CD40 è un recettore della superficie cellulare espresso sulla superficie delle cellule B mature normali e neoplastiche. Mutazioni di CD40 causano la mancata espressione della proteina sulla superf...

Il deficit di CD40L (CD154) è caratterizzato da bassi o assenti livelli nel siero di IgG, di IgA e di IgE, con normali o elevati livelli di IgM. I pazienti affetti sono predisposti ad infezioni ricor...

Il deficit di Uracile-DNA glicolasi (UNG) è caratterizzato da bassi o assenti livelli nel siero di IgG, di IgA e di IgE, con normali o elevati livelli di IgM. I pazienti affetti sono predisposti ad ...

Il Nuclear factor kappaB (NF-kB) è un fattore di trascrizione nucleare presente in tutte le cellule che producono citochine, fattori di crescita, chemochine, molecole di adesione, recettori per tutt...

La capacità di Nf-kB di sopprimere l'apoptosi e di regolare la transizione del ciclo cellulare indica chiaramente che Nf-kB svolge un ruolo importante nell’oncogenesi. Un’aumentata attività di N...

I dettagli del ciclo cellulare variano da organismo ad organismo e secondo la fase di vita dell'organismo stesso. Alcune caratteristiche del ciclo tuttavia, sono universali.I dettagli del ciclo cellul...

L’apoptosi, o morte cellulare programmata, è un processo fisiologico che negli organismi multicellulari mantiene l’omeostasi cellulare ed elimina le cellule difettose o in soprannumero. L’apopt...

I linfociti sono le cellule più importanti del sistema immunitario adattativo o acquisito. Un sistema immunitario efficiente richiede la formazione di linfociti in grado di riconoscere virtualmente t...

I linfociti B sono le cellule deputate alla sintesi e secrezione delle immunoglobuline (Ig), previa trasformazione in cellule altamente specializzate, le plasmacellule

La sindrome di Wiskott-Aldrich è un disordine congenito trasmesso come carattere recessivo laegato al sesso che nella forma classica l si manifesta con diatesi emorragica da piastrinopenia, eczema, ...

La diagnosi di A-T si basa sulla clinica e sulle anomalie di laboratorio nessuna delle quali è tuttavia patognomonica per la malattia. La diagnosi è fondata sulla presenza delle caratteristiche ma...

L’Atassia- Teleangiectasia (A-T) è una malattia sistemica progressiva con interessamento prevalente dle sistemi nervoso e muscolare. Altre manifestazioni comprendono deficit immunitario con infe...

La COVID-19 è principalmente una polmonite virale che, in una minoranza di casi, progredisce ad una malattia sistemica grave, potenzialmente fatale, caratterizzata da febbre resistente al trattament...

Il plasma convalescente, o iperimmune, è stato utilizzato come strumento per fornire una rapida protezione (immunità passiva) a pazienti in gravi condizioni nel corso di numerose epidemie/pandemie, ...

La maturazione della cellula pre-B nel midollo emopoietico richiede la presenza sequenziale di alcune molecole necessarie per l'assemblaggio del BCR, senza il quale la maturazione delle cellule B s...

Circa il 10-15% dei pazienti con agamaglobulinemia di Bruton, o XLA, non soddisfa i criteri clinici e di laboratorio per una diagnosi tipica, pur essendo portatori del gene BTK mutato. In questi casi ...

L’agammaglobulinemia di Bruton è causata dalla carenza di BTK o Bruton’s tyrosine kinase, una tirosinchinasi citoplasmatica che nei linfociti B svolge un ruolo importante nella trasduzione del se...

I pazienti affetti da linfoma folicolare avanzato convivono con il linfoma per anni in buone condizioni di salute, anche senza ricevere terapia. Inoltre, una duratura remissione completa può essere...

I pazienti con linfoma follicolare avanzato asintomatici possono essere, almeno all'inizio, soltanto seguiti con l'osservazione clinica (watch and wait, cioè osserva e attendi la progressione prima ...

Numerosi studi hanno dimostrato che la chemioimmunoterapia aumenta le percentuali di risposte, la durata della PFS e della sopravvivenza globale. La sola chemioterapia non è oggi indicata nella maggi...

Per la diagnosi di linfoma follicolare è richiesta la biopsia chirurgica, con asportazione completa del linfonodo di maggiori dimensioni oppure la biopsia di un sito extra linfonodale coinvolto; l’...

Il trattamento dei pazienti affetti da linfoma follicolare in stadio veramente localizzato (stadio I) è basato principalmente sulla radioterapia, che è in grado di guarire molti pazienti. Invece il ...

I linfomi follicolari sono graduati su una scala da uno a tre secondo il numero di centroblasti ad alto ingrandimento (vedi immagine) . I linfomi follicolari di grado IIIA possono essere considerati d...

I pazienti asintomatici possono essere seguiti in ambulatorio ogni tre mesi per il primo anno e poi ogni tre-sei mesi negli anni successivi fino al momento della diagnosi di progressione clinica. Nei ...

La terapia moderna dei linfomi follicolari sintomatici o ad alto volume tumorale secondo i criteri GELF o BNLI consiste nell’associare un anticorpo monoclonale, rituximab o obinutuzumab, con la ...

L'uso del trapianto di cellule staminali emopoietiche nel linfoma follicolare è materia di controversie e di accesi dibattiti nella comunità scientifica, essendo stato oggetto numerosi studi clinic...

L'uso del trapianto di cellule staminali emopoietico nel linfoma follicolare è stato oggetto di dibattito e di analisi in numerosi studi clinici. Il trapianto di cellule staminali emopoietiche è di...

l linfoma di Burkitt è un raro tipo di linfoma non Hodgkin ad alta aggressività che origina dai linfociti B maturi e ha un tempo di duplicazione cellulare molto breve. Nei primi anni del 20º secolo...

l linfoma di Burkitt è un raro tipo di linfoma non Hodgkin ad alta aggressività che origina dai linfociti B maturi e ha un tempo di duplicazione cellulare molto breve. Da sempre è stata chiara la r...

Il virus di Epstein-Barr (EBV) (o Human Herpesvirus 4, HHV-4) è un herpesvirus che si diffonde per contatto diretto, mediante trasmissione con la saliva. L’infezione primaria inizia nell’orofarin...
La terapia del linfoma di Burkitt si è evoluta negli ultimi decenni adeguandosi di pari passo alla comprensione delle principali caratteristiche biologiche e molecolari della malattia. Essendo un l...
Tutte le cellule tumorali della variante africana del linfoma di Burkitt (con rare eccezioni) contengono copie multiple del genoma del virus di Epstein-Barr (EBV). Il virus persiste innanzitutto nei l...
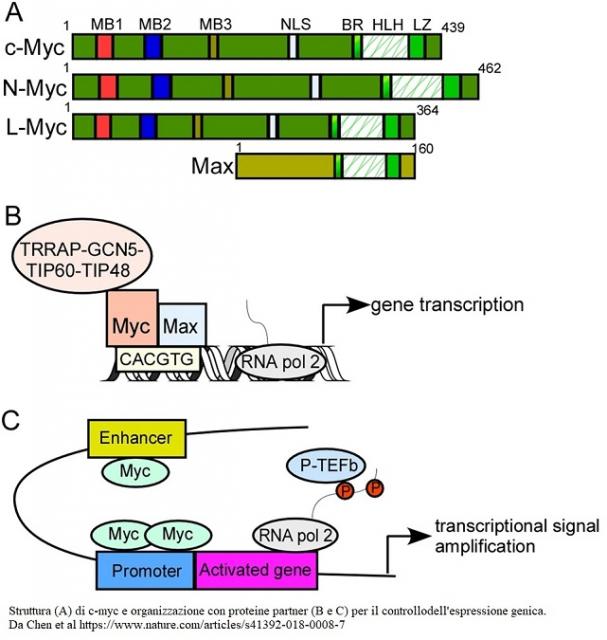
c-myc è un fattore di trascrizione nucleare omologo dell’ oncogène virale Avian Myelocytomatosis o v-c-myc che causa leucemie e sarcomi nei polli. c-myc codifica per una fosfoproteina nucleare con...
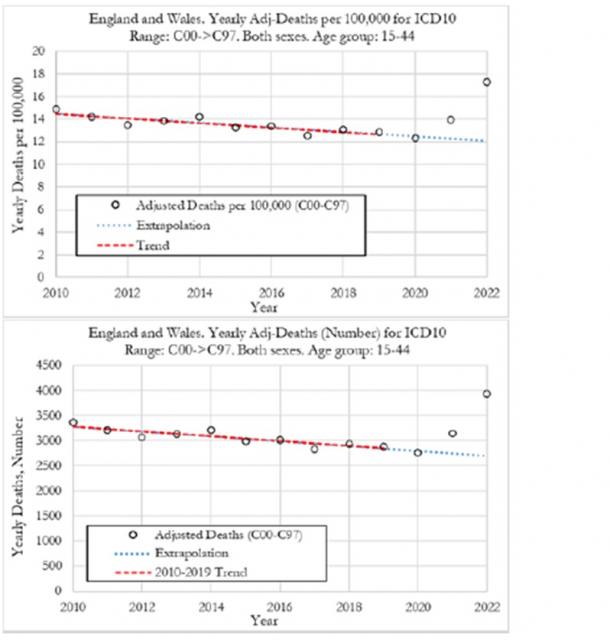
L’uso massiccio e generalizzato, specie nei paesi occidentali, di vaccini a mRNA per contrastare la pandemia COVID-19 da SARS-CoV2 sta sollevando crescenti perplessità circa la loro efficacia e sic...