
Immunodeficienza da deficit di Artemis
Il gene DNA cross-link repair 1C (DCLRE1C) è indispensabile per la ricombinazione dei geni variabili V(D)J dei recettori TCR e BCR sui linfociti T e B e per lo svolgimento della NHEJ che permette la...
Il gene DNA cross-link repair 1C (DCLRE1C) è indispensabile per la ricombinazione dei geni variabili V(D)J dei recettori TCR e BCR sui linfociti T e B e per lo svolgimento della NHEJ che permette la...

I sintomi tipici della SCID sono: le gravi infezioni ricorrenti, soprattutto delle vie respiratorie e del tratto gastrointestinale; la diarrea cronica e la ridotta velocità di crescita. Se non ident...

Le sindromi da iper IgM sono un gruppo di rare immunodeficienze primitive congenite ed ereditarie caratterizzate da: infezioni ricorrenti ad insorgenza precoce, diminuzione o assenza nel siero di IgG ...

La sindrome di Comèl-Netherton (NETH, OMIM*25650) è una rara malattia autosomica recessiva con interessamento della pelle, dei capelli del sistema immunitario caratterizzata dalla triade di ittiosi...

Il deficit di beta-2 microglobulina è stato descritto finora in soli due casi che presentavano una grave immunodeficienza di tipo combinato o SCID..

Le Janus kinasi (JAK) sono delle tirosinchinasi non recettoriali conservate durante la filogenesi e distribuite ubiquitariamente nelle cellule dei mammiferi. Nell’uomo si conoscono 4 membri della fa...

Il deficit di IL6ST può essere causato da mutazioni omozigoti o eterozigoti composite del gene IL6ST, che codifica per la gp130, oppure da mutazioni eterozigoti dominanti negative di un solo allele d...

Le immunodeficienze combinate (CID) sono delle rare sindromi da errori congeniti dell’immunità caratterizzate da gravissimi deficit quantitativi e/o qualitativi dei linfociti T con o senza difetti...

Le mutazioni del gene Protein Kinase, Dna-Activated, Catalytic Subunit (PRKDC ) (OMIM*600899) sono trasmesse come carattere autosomico recessivo e sono causa di una forma di SCID T-B-NK+

La sindrome di Wiskott-Aldrich è un disordine congenito trasmesso come carattere recessivo laegato al sesso che nella forma classica l si manifesta con diatesi emorragica da piastrinopenia, eczema, ...
Il prototipo delle sindromi da Iper IgE è la sindrome di Job o HIES-1, causata da deficit di STAT3 che si manifesta fin dall’infanzia o già al momento della nascita con la caratteristica triade ...

I deficit di fosfoglucomutasi 3 (PGM3) causano una forma di sindrome da iper-IgE (HIES) a trasmissione autosomica recessiva (AR). I deficit congeniti di PGM3 e di altri enzimi coinvolti nella glicos...
ZNF341 è un fattore di trascrizione nucleare di tipo zinc finger che si lega al promoter di numerosi geni, fra i quali STAT3. Mutazioni Loss of Function eterozigoti di ZNF341, un gene precedentemente...

ILgene IL-6R codifica per la subunità alfa del recettore per IL-6..L‘altra subunità del recettore, la gp130 o IL-6ST, permette la trasmissione del segnale originato dall’attivazione di IL-6R all...

Il deficit di TYK2 causa una rara sindrome da immunodeficienza primitiva con predisposizoine alle infezioni da micobatteri, da altri batteri intracellulari e virali. La sindrome da iper-IgE, descritt...

Le mutazioni omozigoti null di CARD11 causano un’immunodeficienza combinata grave (SCID) a trasmissione autosomica recessiva (AR), caratterizzata da: normale numero di linfociti B e T, ma con defi...

La LAD-3, (OMIM 612840), è causata da mutazioni del gene FERMT3 (o KINDLIN3) (OMIM*607901) che codifica per una proteina intracellulare, la Kindlina 3, che interagisce con le β2 integrine. Oltre a...

La LAD-2 è causata da mutazioni nel gene SLC35C1 (OMIM*605881) (Solute carrier family 35 member C1), che è localizzato sul cromosoma 11p11.2. SLC35C1 codifica per la proteina GDP-fucosio transpor...

Il deficit di adesione leucocitaria di tipo 1 (LAD-1) è il più frequente dei tre difetti genetici classificati nel gruppo delle LAD. La LAD-1, OMIM 116290, a trasmissione autosomica recessiva, è ca...

I leucociti migrano dal circolo al sito dell’infiammazione seguendo un gradiente di chemochine provenienti dalla sede dell’infiammazione/infezione in un processo noto come chemiotassi. I fattori c...

La risposta di difesa dell’organismo ai microrganismi esterni è il risultato del concorso di un insieme di sistemi di protezione, comprese barriere fisiche, vari tipi cellulari e mediatori solubili...

L’immunodeficienza combinata severa (Severe Combined Immune Deficiency, SCID) legata al sesso o X-linked è la forma più frequente fra tutte le immunodeficienze primitive combinate. È nota anche...

L’incidenza e la prevalenza delle immunodeficienze primitive sono difficili da stimare per vari motivi, per esempio per la diversa metodologia utilizzata negli studi condotti in paesi differenti ch...

La SCID T-B-NK+ SCID è causata nell’uomo da mutazioni di almeno 5 geni diversi coinvolti nella ricombinazione V(D)J dei recettori di membrana linfocitari, le immunoglobuline (Ig) nei linfociti B ...
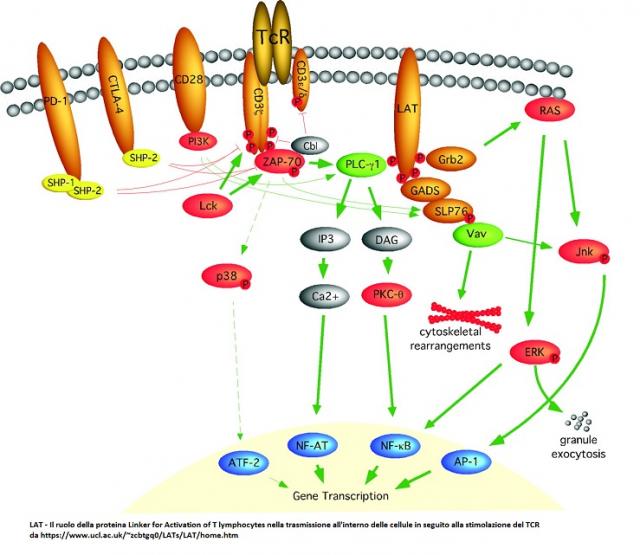
Il legame fra TCR e un suo ligando, un antigene peptidico presentato da una proteina del complesso maggiore di istocompatibilità, innesca cascate di eventi biochimici il cui esito è la stimolazione...

Le immunodeficienze primitive (IP) sono delle rare sindromi genetiche, caratterizzate da anomalo sviluppo di uno o più tipi cellulari del sistema immunitario (linfociti B e T, cellule natural killer,...

Fra le sette coronine note, la 1a è la più importante nei mammiferi e anche quella più studiata. Le mutazioni autosomiche recessive del gene CORO1A causano nei topi e nell'uomo linfocitopenia T di...

Il deficit di IL-7Rα causa un deficit selettivo dei linfociti T ed è una rara causa di SCID T-B+NK. Solo pochi casi sono sstati segnalati, in genere in bambini nati da genitori consanguinei. Non esi...

La sindrome dei linfociti nudi di tipo I, o deficit delle molecole MHC di classe I è un sindrome eterogenea clinicamente e geneticamente. La sindrome può essere causata da mutazioni dei geni TAP1, T...

Cernunnos, dal nome di una divinità celtica protettrice della fertilità, noto anche come Non-homologous end-joining factor 1 (NHEJ1) è un fattore di riparazione del DNA essenziale per il processo d...

Il deficit di molecole MHC II generalmente si traduce in un quadro clinico di SCID, indipendentemente dal difetto molecolare, poiché esse svolgono un ruolo fondamentale nella maturazione e nella f...

La SCID T-B-NK+ SCID è causata nell’uomo da mutazioni di almeno 5 geni diversi coinvolti nella ricombinazione V(D)J dei recettori di membrana linfocitari, le immunoglobuline (Ig) nei linfociti B ...

Il deficit di CD40L (CD154) è caratterizzato da bassi o assenti livelli nel siero di IgG, di IgA e di IgE, con normali o elevati livelli di IgM. I pazienti affetti sono predisposti ad infezioni ricor...

L'anomalia peculiare della SCID è la linfocitopenia, solitamente <1500/µL, che non è però costante [6]. Tuttavia, alla nascita numero assoluto di linfociti <2000/µL, deve far sorgere il sospe...

Riassunto Mutazioni del gene adenosina deaminasi causano accumulo substrati e metaboliti tossici per le cellule, fra quali adenosina, desossiadenosina e desossi-ATP che inducono danni cromosomici b...

Riassunto Mutazioni del gene PNP (OMIM*164050) causano accumulo substrati e metaboliti tossici per le cellule. soprattutto per quelle del sistema immunitario, fra quali adenosina, desossiadenosina...

Il deficit di adenilato chinasi 2 (AK2) causa la disgenesia reticolare, un’immunodeficienza combinata grave di tipo SCID T-B-NK- associata ad agranulocitosi e sordità neurosensoriale bilaterale....

Il deficit di adenosina deaminasi indotta dall'attivazione (AICDA) è caratterizzato da bassi o assenti livelli nel siero di IgG, di IgA e di IgE, con normali o elevati livelli di IgM. I pazienti aff...

Il CD40 è un recettore della superficie cellulare espresso sulla superficie delle cellule B mature normali e neoplastiche. Mutazioni di CD40 causano la mancata espressione della proteina sulla superf...

Il deficit di Uracile-DNA glicolasi (UNG) è caratterizzato da bassi o assenti livelli nel siero di IgG, di IgA e di IgE, con normali o elevati livelli di IgM. I pazienti affetti sono predisposti ad ...

La fase acuta delle infezioni da SARS-CoV e SARS-CoV-2 nell'uomo è associata a una marcata linfocitopenia e a una profonda compromissione delle risposte immunitarie e infiammatoria. A differenza di...

I linfociti T maturi riconoscono gli antigeni per mezzo di un eterodimero variabile (αβ o γδ), della membrana di superficie noto come TCR o recettore delle cellule T. Nell'uomo, le subunità d...

CD45 è codificata dal gene PTPRC (Protein Tyrosine Phosphatase Receptor Type C). CD45 esiste in diverse isoforme, originate da splicing alterativo degli esoni 4,5, 6. CD45 è una molecola recettorial...

L’Atassia- Teleangiectasia (A-T) è una malattia sistemica progressiva con interessamento prevalente dle sistemi nervoso e muscolare. Altre manifestazioni comprendono deficit immunitario con infe...

La diagnosi di A-T si basa sulla clinica e sulle anomalie di laboratorio nessuna delle quali è tuttavia patognomonica per la malattia. La diagnosi è fondata sulla presenza delle caratteristiche ma...

Pur trattandosi di malattie rare, con il diffondersi della pandemia da SARSCoV 2 ancora in corso, è possibile che un numero crescente di pazienti affetti da immunodeficienza primitiva (IP) sviluppi...

L’agammaglobulinemia di Bruton è causata dalla carenza di BTK o Bruton’s tyrosine kinase, una tirosinchinasi citoplasmatica che nei linfociti B svolge un ruolo importante nella trasduzione del se...

Circa il 10-15% dei pazienti con agamaglobulinemia di Bruton, o XLA, non soddisfa i criteri clinici e di laboratorio per una diagnosi tipica, pur essendo portatori del gene BTK mutato. In questi casi ...

La maturazione della cellula pre-B nel midollo emopoietico richiede la presenza sequenziale di alcune molecole necessarie per l'assemblaggio del BCR, senza il quale la maturazione delle cellule B s...

Il gene DNA ligasi IV, o LIG4, proteina essenziale per la giunzione terminale non omologa del DNA, nonhomologous DNA end joining (NHEJ), il processo che opera il ricongiungimento delle estremità del...